

От перебора к пониманию: почему химии нужен новый тип ИИ
Современная фарма живет в парадоксе: потенциально полезных молекул очень много, а времени и лабораторных ресурсов всегда мало. По оценкам, пространство малых молекул, которые теоретически могут стать лекарствами, лежит в диапазоне от 10^20 до 10^60. Даже нижняя граница настолько огромна, что классический подход «синтезируй и проверь» перестает работать в разумные сроки.
Именно здесь появляется направление, которое развивает команда MIT под руководством Коннора Коули: использовать ИИ не как «генератор случайных идей», а как систему, которая учитывает химическую логику — форму молекул, механизмы реакций и физические ограничения.
Кто такой Коннор Коули и почему его подход важен
Коннор Коули работает на стыке химической инженерии и компьютерных наук в MIT (Chemical Engineering + EECS + Schwarzman College of Computing). Его исследовательская линия проста по формулировке и сложна по сути: построить вычислительные модели, которые помогают:
- анализировать гигантские химические пространства,
- проектировать новые молекулы с нужными свойствами,
- предсказывать реалистичные пути синтеза.
Ключевой тезис Коули: модели должны быть «приземлены» в химии так же, как рассуждает опытный химик. Это важный сдвиг для всего AI for Science: точность растет не только от большего объема данных, но и от встроенных научных ограничений.
Почему «умная генерация» лучше «слепой генерации»
Многие генеративные модели в науке работают как очень быстрый мозговой штурм: они предлагают варианты, но не всегда понимают, можно ли их действительно получить в колбе. В химии это критично. Кандидат может выглядеть красиво на экране, но быть непригодным в синтезе или нестабильным.
Подход MIT пытается решить эту проблему через химическую интуицию в модели. Аналогия: если обычная модель похожа на человека, который придумывает рецепты, не умея готовить, то модель Коули похожа на шефа, который знает технику, совместимость ингредиентов и последовательность шагов.
Два знаковых инструмента: ShEPhERD и FlowER
ShEPhERD — модель, которая оценивает кандидаты в лекарства по тому, как их 3D-форма может взаимодействовать с белком-мишенью. Это важно, потому что в медхимии «подходит по форме» часто не менее важно, чем «подходит по формуле».
FlowER — генеративная модель для предсказания продуктов реакции. Ее принципиальное отличие: в архитектуру заложены фундаментальные ограничения, включая закон сохранения массы, а также проверка правдоподобия промежуточных стадий реакции.
Результат, о котором сообщают исследователи: такие ограничения повышают точность прогноза по сравнению с более «свободными» моделями.
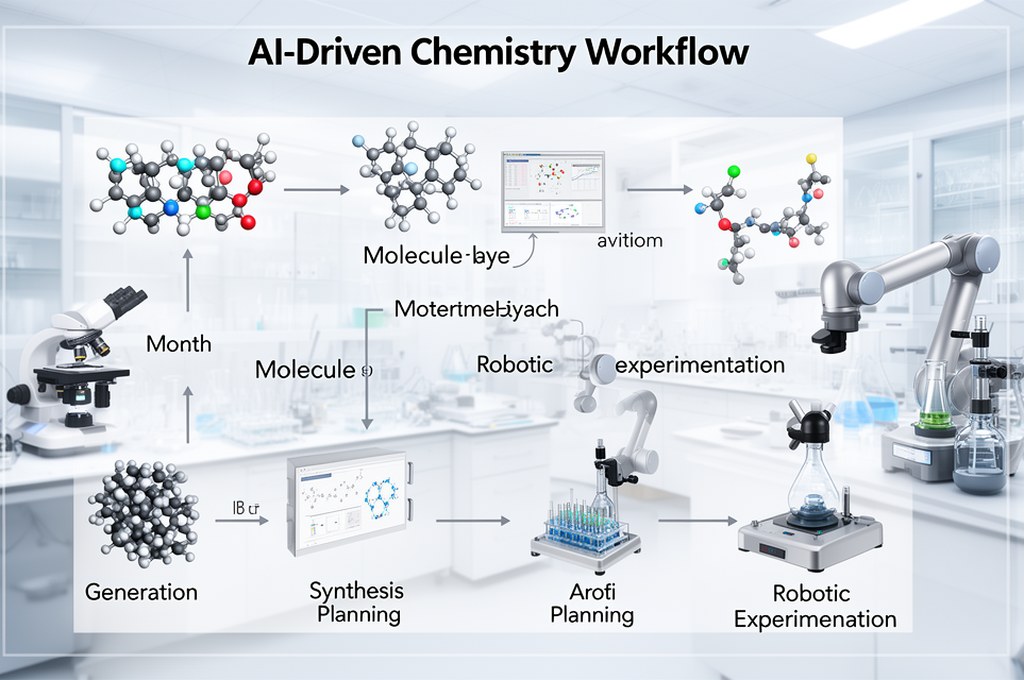
Что здесь действительно нового для индустрии
1. Переход к physics-informed и mechanism-aware AI
В последние годы ИИ в химии часто критиковали за «галлюцинации синтеза». Работа в духе Коули задает тренд: модель должна соблюдать научные законы, а не только статистические паттерны.
2. Снижение стоимости ошибки на ранних этапах
В drug discovery самая дорогая ошибка — поздняя. Если модель раньше отсеивает неперспективные молекулы и неосуществимые маршруты, компании экономят месяцы и миллионы долларов.
3. Рост роли гибридных команд
Этот кейс MIT показывает, что будущее не за «чистыми» AI-командами или «чистыми» химиками, а за смешанными группами: медхимия + ML + автоматизация лабораторий + оптимальный дизайн экспериментов.
Сравнение подходов: классический pipeline vs AI с химической интуицией
| Критерий | Классический подход | AI с химическими ограничениями |
|---|---|---|
| Поиск кандидатов | Ограниченный, медленный перебор | Широкий и быстрый скрининг |
| Проверка синтезируемости | Часто поздно в pipeline | Частично учитывается на этапе генерации |
| Учет 3D-взаимодействий | Точечно, вручную и дорого | Системно, в модели (пример ShEPhERD) |
| Риск «нереальных» предложений | Ниже, но медленно | Снижается за счет механизмов и законов (пример FlowER) |
| Скорость итераций | Недели и месяцы | Дни и часы для виртуальных циклов |
Практические последствия для фармы и биотеха
- Big Pharma получает инструмент ускорения preclinical-этапов и расширения химического разнообразия.
- Биотех-стартапы могут конкурировать эффективнее, потому что ИИ частично компенсирует дефицит «мокрой» инфраструктуры.
- CRO и CDMO получают спрос на автоматизированный синтез и цифровые платформы планирования реакций.
- Университеты будут активнее перестраивать программы, где химики учатся ML, а ML-инженеры — реакционной механистике.
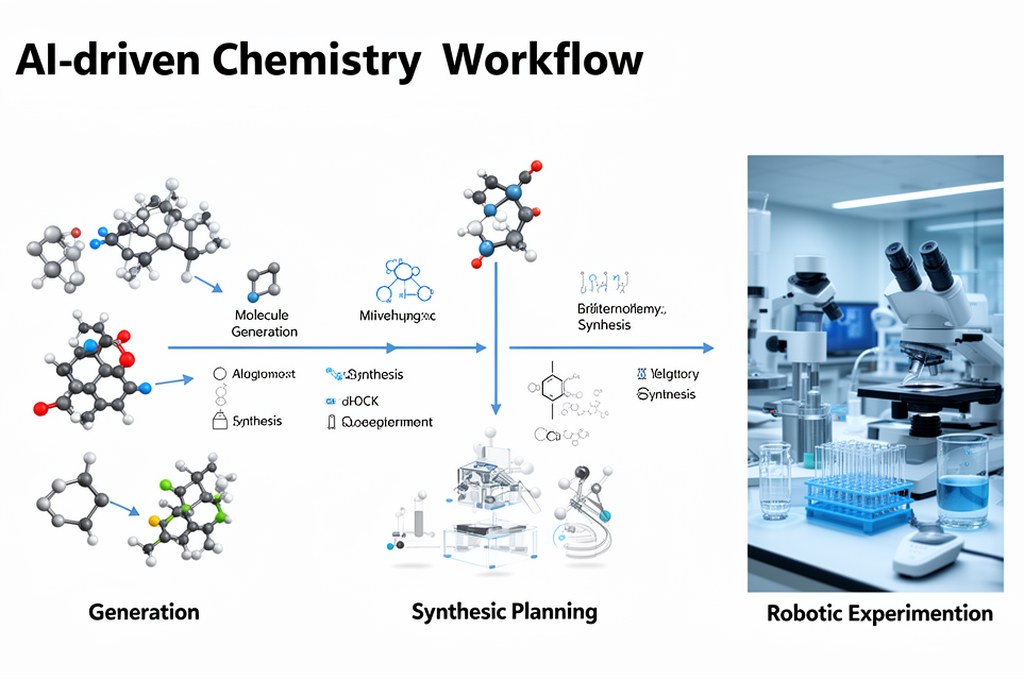
Ограничения: что пока не решено
Важно не переоценивать прогресс. Даже лучшие модели:
- зависят от качества обучающих данных,
- могут хуже переноситься на редкие классы реакций,
- не заменяют экспериментальную валидацию, а лишь оптимизируют ее.
Иначе говоря, ИИ пока не «автономный химик», а очень сильный ко-пилот исследователя.
Прогноз на ближайшие 3-5 лет
С высокой вероятностью мы увидим интеграцию трех слоев в единый цикл:
- генерация молекул с учетом биологической цели,
- ретросинтез и оценка реализуемости маршрута,
- роботизированная лабораторная проверка с автоматическим обновлением модели.
Когда этот контур замыкается, наука начинает работать как самообучающаяся производственная система. Именно к этому, по сути, ведет линия исследований MIT.
Вывод
Материал MIT о работе Коннора Коули важен не как «еще одна новость про ИИ», а как сигнал о зрелости направления. Следующий этап AI в химии — это не просто большие модели, а модели, которые уважают законы природы и логику реакций. Для индустрии это означает ускорение поиска лекарств, более рациональное использование лабораторных ресурсов и постепенный переход к действительно инженерному, воспроизводимому drug discovery.







